For U.S. Healthcare Professionals
For U.S. Healthcare Professionals
If you click “Continue” below, you will leave the current site and be taken to a site maintained by a third party that is solely responsible for its content. Amgen provides this link as a service to website visitors. Amgen is not responsible for the privacy policy of any third-party websites. We encourage you to read the privacy policy of every website you visit.


Cholesterol plays an important role in many physiological functions, but it can become harmful at excessive plasma levels and it’s a key risk factor for CV events. Cholesterol is mainly produced by the liver, which also regulates plasma LDL through LDL-receptor (LDLR)-mediated clearance.
LDL Circulation4
Dietary cholesterol and cholesterol produced de-novo are transported in the blood by LDL and other apolipoprotein B (apoB)-containing lipoproteins.

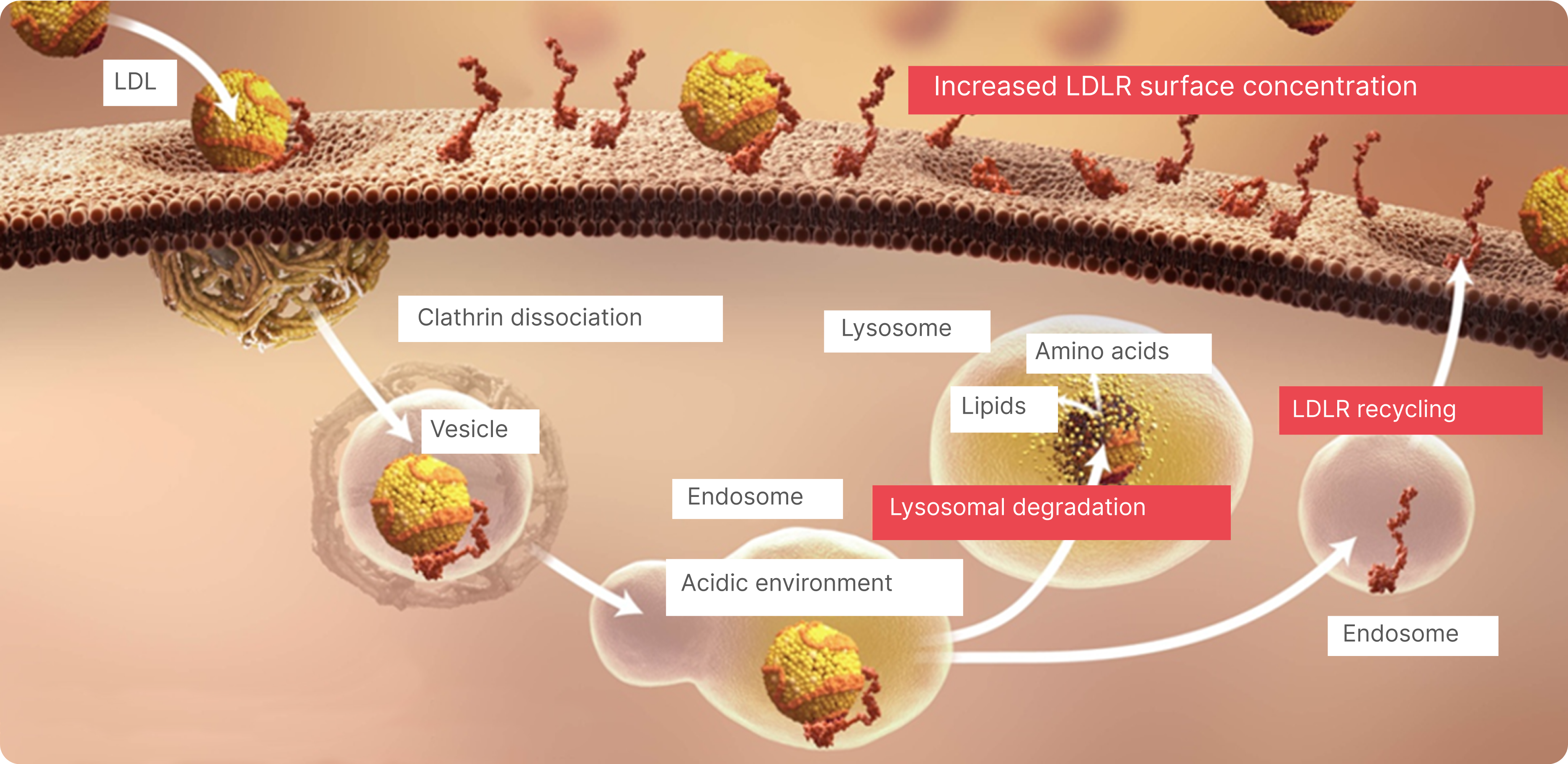
LDL Uptake in the Liver1,4
ApoB-containing lipoproteins bind to LDLRs on the surface of hepatic cells and receptor-bound lipoproteins are internalized through endocytosis (the cell absorbs LDL).

LDL Clearance1
Internalized LDLRs dissociate from apoB-containing lipoproteins and are recycled back to the cell surface while LDL is hydrolyzed in lysosomes.
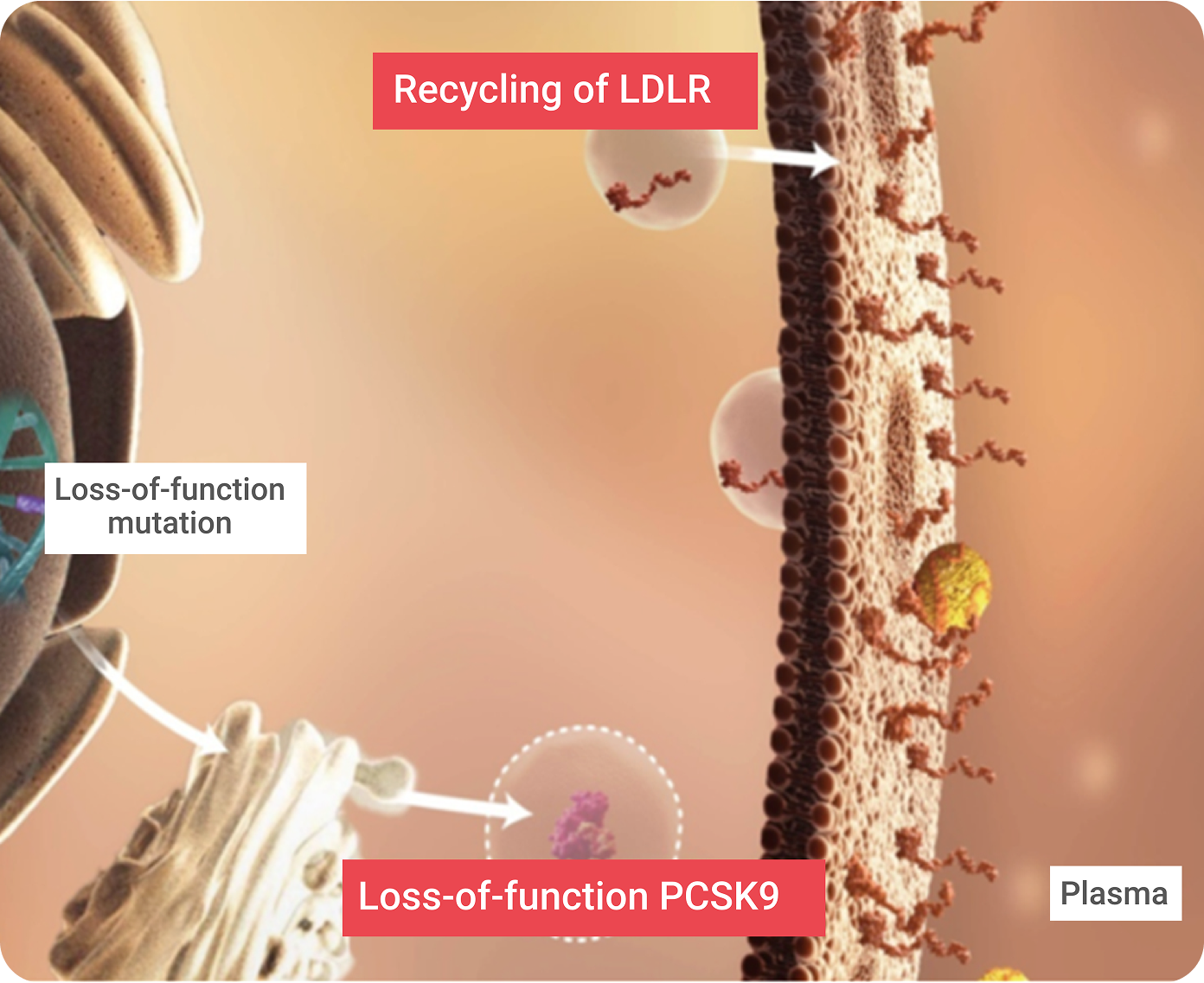
Recycling of LDLRs Enables Efficient Clearance of LDL Particles From the Blood1,5,6
An estimated 70% of systemic LDL-C is cleared from the circulation through the hepatic cell LDLRs. Complex feedback loops maintain constant levels of intracellular LDL-C, despite changes in circulating LDL-C levels.
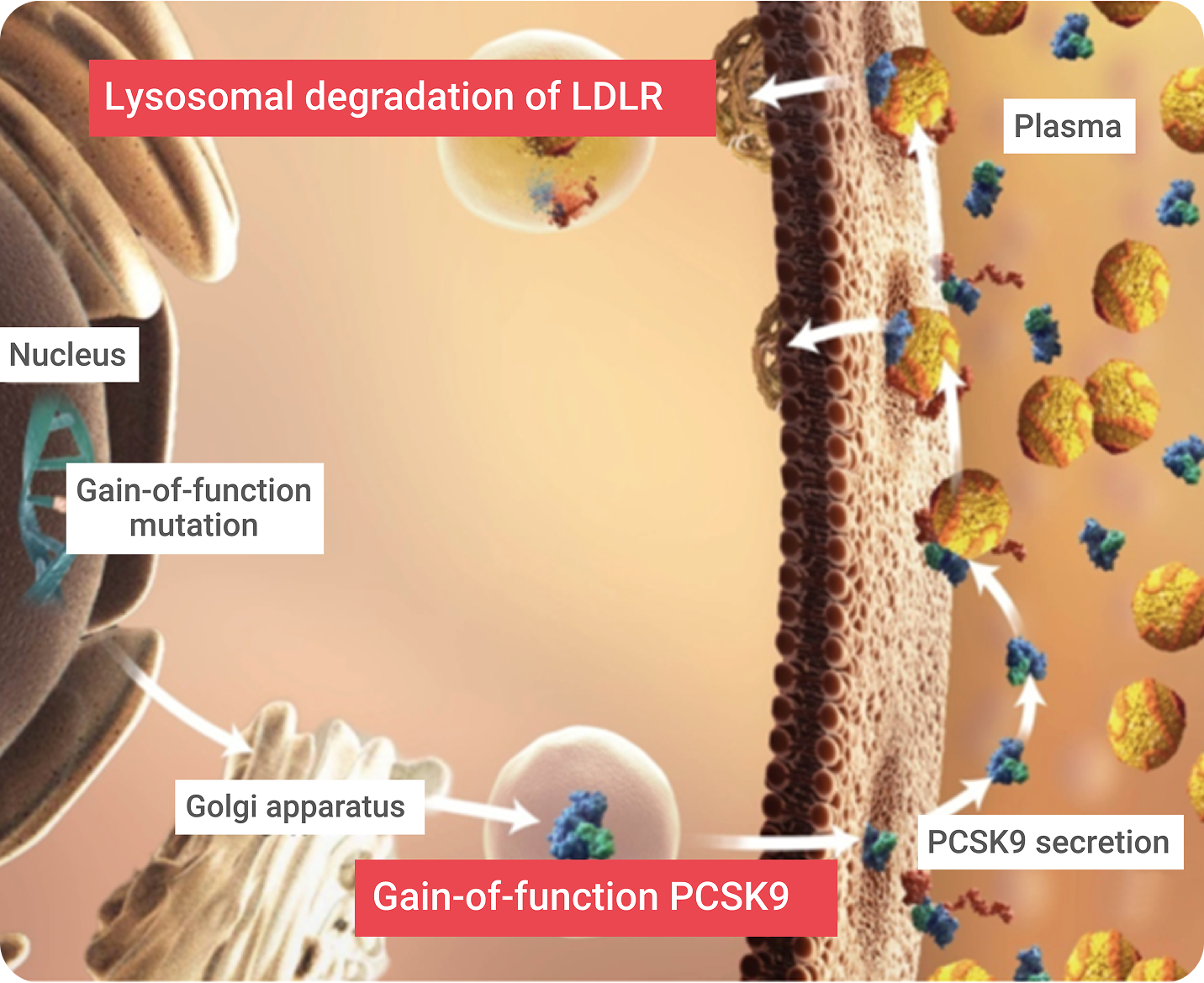
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a member of the mammalian proprotein convertase family of secretory serine endoproteases. PCSK9 functions as a molecular chaperone, binding to an LDLR/LDL-C complex and targeting it for lysosomal degradation once the LDLR/LDL-C complex has been pulled into the hepatic cell. This helps achieve LDL-C homeostasis and controls the amount of LDLRs on the hepatic cell surface. PCSK9 directs the degradation of LDLR via two separate pathways:
Gain-of-function (GOF) mutations5
result in fewer LDLRs, which causes high serum LDL-C (associated with familial hypercholesterolemia [FH]).
Loss-of-function (LOF) mutations5
result in more LDLRs, which is associated with lower serum LDL-C.
FH is an autosomal co-dominant disorder most commonly caused by the following mutations:2,8

If left untreated, people with FH reach elevated levels of LDL-C and develop CHD early in life.9
Heterozygous FH (HeFH) affects between 1 in 200 to 250 people and is characterized by marked hyperlipidemia (LDL-C ≥ 190 mg/dL), premature development of atherosclerosis, and coronary artery disease.9,10
Percentage of people with untreated HeFH who will experience a coronary event before the age of 6011

~50% MEN

~30% WOMEN
Homozygous FH (HoFH) affects 1 in 1,000,000 people, is characterized by LDL-C > 500 mg/dL, and presents a much more severe phenotype than people with HeFH. People with HoFH have plasma LDL-C 2x higher than HeFH, which puts them at risk of premature development of atherosclerosis and coronary artery disease.2,9,12

On average, people with FH who do not receive treatment reach the cumulative LDL-C burden sufficient for the development of CHD 20 years earlier than people without FH.9
CHD = coronary heart disease; FH = familial hypercholesterolemia.